
It’s been quite a year: COVID-19, a change in administration and a year of transition for FDA digital health policy. In light of all the change and uncertainty, many digital health companies are struggling to decide upon a regulatory strategy for 2021.
My advice? If at all possible, stay away from FDA. They have enough to do right now. The people I know at FDA have been working 12- and 15-hour days most of the year. They’ve been doing incredible work, both on the review side and the policy side, when it comes to COVID-19. In fact, if you haven’t already done so, I would recommend that you send anyone you know at FDA a holiday card. And thank them for their service.
But back to business. You need to stay away from FDA if you can. We need to confront reality. Here are three reasons why you should try to stay away.
HIMSS20 Digital
1. There is no benefit to being FDA regulated if you can avoid it
As a preliminary matter, let me explain what I mean by “if you can avoid it.” Everyone knows the difference between tax evasion and tax avoidance. Tax evasion is lying about something in order to avoid paying tax. Tax avoidance is prudent planning, for example, to fund a Roth IRA. I am advocating FDA avoidance, not FDA evasion.
FDA’s regulation fundamentally revolves around claims made about products. Typically, for huge number of digital products, there are claims FDA would not regulate and there are claims FDA would. I’m strongly recommending that you consider limiting yourself to making unregulated claims for the near future.
The path to regulatory authorization for many digital health products is long, unpredictable and expensive
Let’s look at the data through September 30, the end of the government’s fiscal year 2020.
Novel technologies, and those include many of the new digital health products, are not eligible for pre-market notification because there is no predicate device already on the market. As a consequence, such products must be submitted in a de novo application.
But the de novo process is not a place you want to go. It is highly unpredictable for the simple reason that there are neither guidance documents nor a general path to follow specifically for the new device.
FDA gets to examine the device’s fundamental safety and effectiveness, as opposed to substantial equivalence, so FDA asks many wide-ranging questions. Also, clinical trials are typically required, and those trials are costly.
That means the process is also uncertain from an outcome standpoint. Consider this data on the chances of success with a de novo submission.
Look at the rate of granted decisions. It’s generally under half. Now compare that with similar data for the 510(k) process.
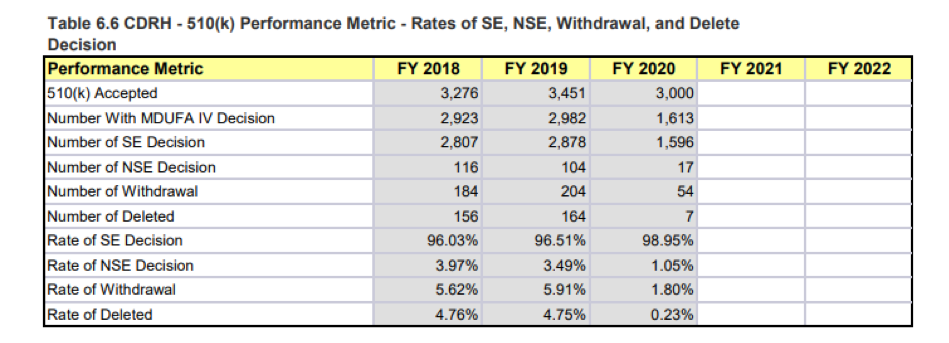
The success rate is typically well north of 95%. Quite a difference from de novo.
And the process is slow, as shown below. Remember, much of these data are before COVID-19.
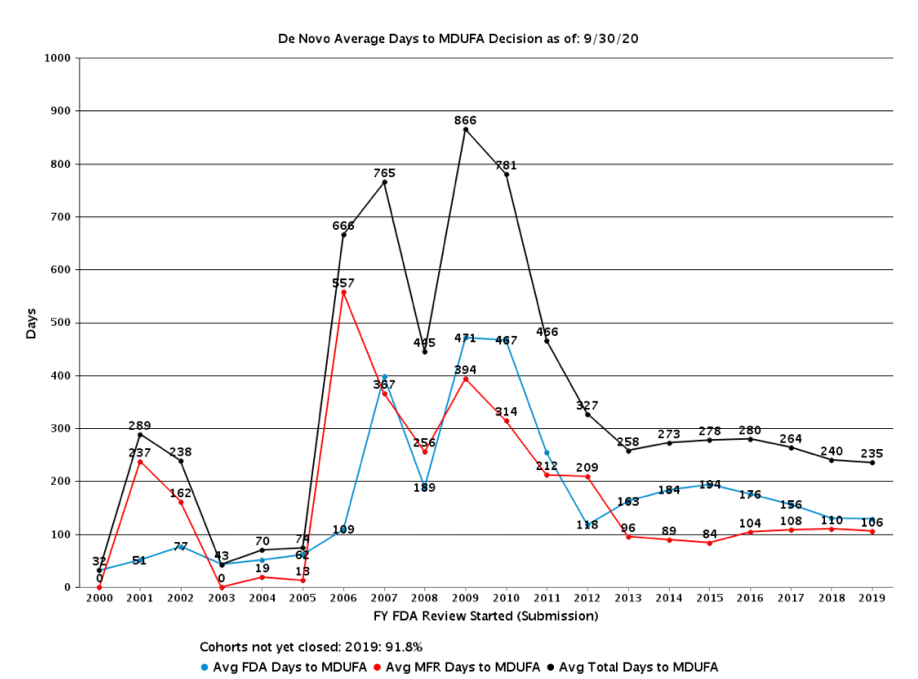
In a way, recent years look almost good compared to 10 years ago. But that’s only because 10 years ago the data were horrendous. This still means that review cycles are well above 200 days. Given COVID-19, those numbers are almost sure to spike up when next reported.
In comparison, the 510(k) numbers are more like half that time.
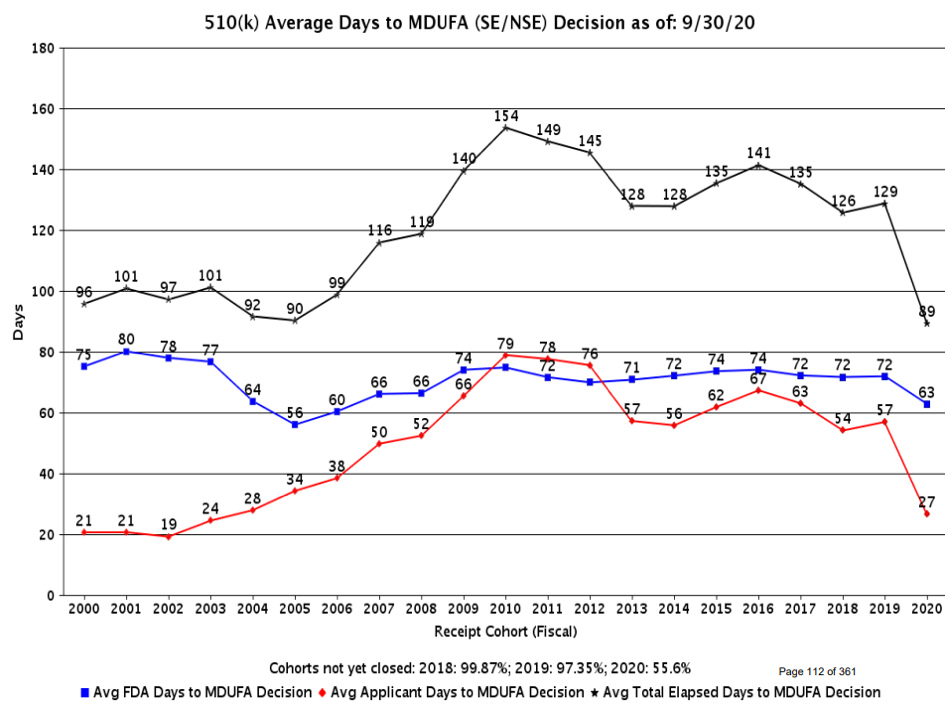
Over the last decade or so, the 510(k) times have also been much more predictable. Please note that for fiscal year 2020, much of the data are still not tabulated.
On the whole, if your digital product is unique enough that the 510(k) pathway is not available, and you are forced to go the de novo route, even before COVID-19 struck, the process was uncertain, lengthy and expensive, given the evidentiary requirements typically imposed by the agency.
FDA clearance may not produce a marketing or reimbursement benefit
I will not spend much time on this point, because it isn’t unique to this year, and frankly it deserves an article unto itself. But I’ve had several clients walk through my door saying that they wanted to be FDA regulated because they felt like it was sort of like the Good Housekeeping Seal of Approval, that it would lead to better acceptance among customers and payers.
I have not seen very good data on this, and you can imagine the difficulty of doing a controlled trial where randomly some companies go get FDA clearance or approval and others don’t. It’s something that’s going to be open to debate, because the data just don’t exist.
All I can tell you is, anecdotally, the clients I know who believed this to be true when they pursued FDA clearance or approval did not believe it to be true once they got FDA clearance or approval. It didn’t lead, certainly not by itself, to any substantial increase in revenue.
You’ll have to decide on your own. But if you are going to invest substantial money securing FDA clearance or approval, you really ought to have evidence that the effort will be worth it.
FDA clearance does not protect companies from competition that breaks the rules
FDA enforcement in digital health has been almost nonexistent for years. At one point, it got so bad that I testified before Congress about a company that FDA was turning a blind eye toward, and it took that for FDA to send an enforcement letter.
FDA publishes its Warning Letters on its website. At the time of this writing, so far in 2020 there have been 27 Warning Letters issued by the Center for Devices and Radiological Health (CDRH). Perhaps not surprisingly, the most recent 16 letters all involved exaggerated or unproven claims related to COVID-19. Before that, the letters were a mishmash of issues from medical device reporting to quality system problems. But not a single one of them – not one – involved unapproved claims for a digital health product.
Now you might be saying that obviously COVID-19 shifted FDA’s priorities. But the prior years were no different. FDA has not been sending Warning Letters to apps or other digital health products that are not FDA approved as required.
The center does use another enforcement vehicle called an “It Has Come to Our Attention Letters.” These are polite enforcement letters that the agency uses when they think a violation was unintentional by someone who just didn’t know that their product is FDA regulated. FDA does not put all of these letters on their website, so we don’t have a database to search. They do, however, put some of their letters on, and only one relates to a digital health product. Indeed, it addresses the product about which I testified.
Here’s the thing. Violations of FDA law by digital health products are rampant. FDA might say that they don’t have the resources to pursue all of them, but does that mean pursuing none of them? Further, when it comes to resources, I could hire a high school intern, pay her 20 bucks an hour, and she could easily identify a dozen violations per hour for quite a few hours. It just isn’t that hard.
So why isn’t FDA pursuing these companies? It’s a good question, and you ought to ask FDA. When I have, it seems apparent to me that FDA is very concerned politically about looking as though it is anti-innovation. If that’s the case, there’s a pretty simple answer. Change the law. Legalize this stuff. What I object to is having a law on the books that’s unenforced and only followed by ethical companies.
The companies I know in the digital health space that have taken the time to go through the de novo process have been very disappointed that FDA has not kept up its end of the deal by then enforcing the regulatory requirements against companies that would try to go directly to market with the same claims but without FDA clearance. Make no mistake, many companies are struggling competitively because they spent quite a bit of time and money going through the FDA review process, only then to compete with companies not complying with FDA requirements.
In the end, it’s all about the patient, and my fear is that the reputable companies will go out of business and only the disreputable ones will survive. That will not help the patient.
2. COVID-19 has made the process worse
Before COVID-19, based on everything I just said, I would have to say that there’s little benefit to going to FDA for clearance or de novo review if it can be avoided. Then along came COVID-19, and the disease managed to make the process much worse. Here’s how.
CDRH has received well over 3,000 Emergency Use Authorization requests.
As of the middle of September, here are the exact numbers:
- 1,734 pre-EUAs
- 3,040 EUAs
And the work has not let up. With the resurgence of COVID-19, many companies that didn’t get their submissions in for the first wave have chosen now to pursue the second wave.
The problem with that is that it’s all on top of the normal workload, and it doesn’t come with user fees. So it sucks up resources without replenishing them. I’ve talked to some of the FDA leaders, and it appears that the normal workload hasn’t dwindled during 2020. So those EUAs are not instead of normal submissions, but on top of normal submissions.
Further, CDRH published 26 guidance documents related to COVID-19. That policy work obviously took many hours.
The net impact is some branches of CDRH are now refusing to meet with companies
As I understand it, and as you might guess, the brunt of this disruption has occurred in certain offices within CDRH. Those three offices are OHT1 (responsible for anesthesia and respiratory devices), OHT4 (responsible for personal protective equipment, including N95 respirators, facemasks and decontamination systems) and OHT7 (responsible for COVID-19 tests).
Those branches and others have largely shut off all pre-submission meetings, because they don’t have time. That means that if you proceed with the submission, you will need to proceed in the dark without FDA feedback on your planned approach. I’m not blaming FDA. I would do the same thing. It’s that darn COVID-19.
And it’s only natural for submissions to suffer. I submitted an EUA for an important COVID-19 public health device on October 29, and other than an initial review for completeness, I’ve heard nothing from FDA. It’s a shame, because the device in my opinion would be a very important tool in combating the spread of COVID-19.
Duration of this disruption?
If we look at past pandemics, typically the HHS Secretary maintains the emergency declaration for perhaps a year after the number of infections goes down. This is so that the emergency authorization tools remain available in case of a flare up. So if the U.S. gets COVID-19 under control say, in the fall of 2021, it’s likely that the emergency declaration would continue until perhaps the fall of 2022.
The backlog at FDA is not likely to go away anytime soon. The backlog itself is growing, and many of the companies that are pursuing EUAs will then want to get a conventional clearance or approval toward the end of the emergency. We have to anticipate this regulatory environment continuing for perhaps 18-24 months.
3. FDA policymaking is not likely to improve the regulatory environment anytime soon
The new Administration will not help things
I don’t say that as a Democrat or Republican. I say that as an optimist. I consider myself optimistic, because, in my 35 years of observing FDA, I don’t believe that the agency is terribly political. And that ought to be a comfort.
We really shouldn’t want a science-based regulator to be political, drifting significantly with political currents. We should want science to carry the day. And largely it does. New administrations, once they get up and running, poke and prod around the edges, but the rank-and-file at FDA generally continue to do what they always do.
CDRH’s policymaking in digital health is distracted
I say that for a couple of reasons. First, obviously the policymaking apparatus has been focused on COVID-19, because digital health offers remarkable benefits to the healthcare system in a time of a pandemic. Telemedicine in some ways runs on digital health. In the last year, digital health innovators have come up with new ways to use technology in the hands of patients to produce important new diagnostic information, and even deliver therapies remotely.
Further, with the launch of the new Digital Health Center of Excellence, FDA has been trying to recruit talent in such areas as artificial intelligence. But the problem is, budgets were already tight, and COVID-19 has upended those budgets. FDA, as I understand it, is having a difficult time competing for talent in this space in the marketplace.
Finally, I’m afraid that CDRH is distracted by the shiny new thing. There is a group of people at FDA who are really excited about the pre-certification pilot program, notwithstanding the fact that it requires statutory authority, and they have none. But they’re not letting that small detail stop them.
They are working down in the weeds to try to develop the nuances of a pre-certification program that Congress has not authorized. And there are big, controversial issues with regard to the high-level architecture of the program.
FDA is proposing a program where it matters more who you are than what you can do. It would favor the entrenched over the startup. It’s hard to understand how disadvantaging startups in the medical device industry would be good for patients or frankly for industry.
But the other sea-change is that industry would have to accept much deeper and more intrusive post-market regulation. In exchange for precertification, the agency is asking for what would amount to daily, intrusive oversight of marketed products. When the agency can’t handle the work it already has, it’s hard to understand how that would be wise.
FDA is dumping a huge amount of time into trying to work out certain details, apparently under the lobbying strategy that, when it comes to convincing Congress to authorize the program, Congress will feel they have to support it simply because FDA has invested so much time developing it.
Hopefully, though, when the issue gets to Congress, the legislators will take on the much more important issues of whether it is smart to convert the process from regulating devices to regulating companies, and whether it is wise policy to give the agency Big Brother status, including the ability to monitor industry’s moves on a daily basis.
The reason this distraction is such a problem is that there are things that FDA could be doing now that would have a huge impact on digital health. One of them is actually implementing their April 2019 concept paper on artificial intelligence and machine learning. Many folks are very excited about the concepts floated in that paper, and would love to see a draft guidance document implementing them. But alas, FDA’s attention is elsewhere.
Conclusion
Of course, it isn’t always possible to avoid FDA if there is a market that the company truly wants to go after that inherently involves FDA regulation. But typically there are slightly less ambitious claims that a company could make and avoid the requirement of FDA review. Likely throughout 2021 and into 2022, that unregulated pathway will almost certainly be more attractive.
 About the Author: Bradley Merrill Thompson is a member of the firm at Epstein Becker & Green, P.C. There, he counsels medical device, drug and combination-product companies on a wide range of FDA regulatory, reimbursement and clinical trial issues. The opinions in this piece are Thompson’s and don’t necessarily reflect the opinions of MobiHealthNews or HIMSS.
About the Author: Bradley Merrill Thompson is a member of the firm at Epstein Becker & Green, P.C. There, he counsels medical device, drug and combination-product companies on a wide range of FDA regulatory, reimbursement and clinical trial issues. The opinions in this piece are Thompson’s and don’t necessarily reflect the opinions of MobiHealthNews or HIMSS.